Set operations on BED files are an important part of bioinformatics analysis. Because the output of different set operations can vary widely, this article records the basic behavior of common operations.
The test environment in this article is bedtools(v2.31.1). Process substitution is used in the examples so that no temporary test files need to be created. The format is: intersectBed -a <(echo -e "chr1\t100\t200") -b <(echo -e "chr1\t150\t300")
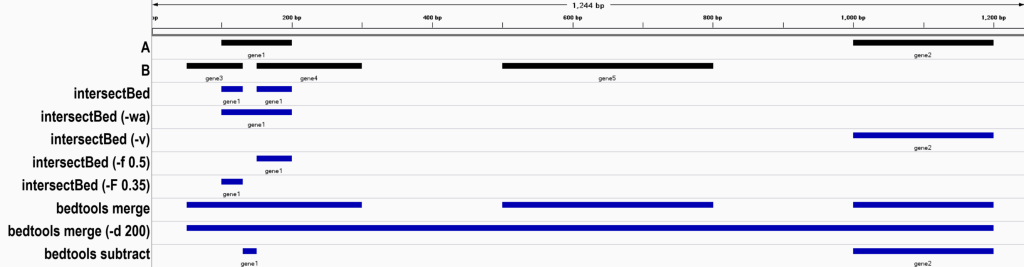
1. Default operation: output intervals in A that overlap B.
# Command
intersectBed -a <(echo -e "chr1\t100\t200\tgene1\nchr1\t1000\t1200\tgene2") -b <(echo -e "chr1\t50\t130\tgene3\nchr1\t150\t300\tgene4\nchr1\t500\t800\tgene5")
# Result
chr1 100 130 gene1
chr1 150 200 gene12. Keep the complete original A record, even when only part of it overlaps.
Parameter: -wa (write original A entry)
# Command
intersectBed -a <(echo -e "chr1\t100\t200\tgene1\nchr1\t1000\t1200\tgene2") -b <(echo -e "chr1\t50\t130\tgene3\nchr1\t150\t300\tgene4\nchr1\t500\t800\tgene5") -wa
# Result
chr1 100 200 gene1
chr1 100 200 gene13. Output intervals in A that overlap B and append the complete overlapping B record.
Parameter: -wb
# Command
intersectBed -a <(echo -e "chr1\t100\t200\tgene1\nchr1\t1000\t1200\tgene2") -b <(echo -e "chr1\t50\t130\tgene3\nchr1\t150\t300\tgene4\nchr1\t500\t800\tgene5") -wb
# Result
chr1 100 130 gene1 chr1 50 130 gene3
chr1 150 200 gene1 chr1 150 300 gene44. Keep the complete original A record and the complete overlapping B record.
Parameters: -wa -wb
# Command
intersectBed -a <(echo -e "chr1\t100\t200\tgene1\nchr1\t1000\t1200\tgene2") -b <(echo -e "chr1\t50\t130\tgene3\nchr1\t150\t300\tgene4\nchr1\t500\t800\tgene5") -wa -wb
# Result
chr1 100 200 gene1 chr1 50 130 gene3
chr1 100 200 gene1 chr1 150 300 gene45. Keep the complete original A record and the complete overlapping B record, and output the overlap length.
Parameter: -wo (write overlap)
# Command
intersectBed -a <(echo -e "chr1\t100\t200\tgene1\nchr1\t1000\t1200\tgene2") -b <(echo -e "chr1\t50\t130\tgene3\nchr1\t150\t300\tgene4\nchr1\t500\t800\tgene5") -wo
# Result
chr1 100 200 gene1 chr1 50 130 gene3 30
chr1 100 200 gene1 chr1 150 300 gene4 506. Keep all complete original A records and complete overlapping B records. For A records without overlap in B, fill missing fields with “-1” or “.”, and output the overlap length at the end.
Parameter: -wao (Write All and Overlap)
# Command
intersectBed -a <(echo -e "chr1\t100\t200\tgene1\nchr1\t1000\t1200\tgene2") -b <(echo -e "chr1\t50\t130\tgene3\nchr1\t150\t300\tgene4\nchr1\t500\t800\tgene5") -wao
# Result
chr1 100 200 gene1 chr1 50 130 gene3 30
chr1 100 200 gene1 chr1 150 300 gene4 50
chr1 1000 1200 gene2 . -1 -1 . 07. Keep the complete original A record. The effect is the same as -wa, but duplicate records are removed.
Parameter: -u (write original A entry once if any overlap)
# Command
intersectBed -a <(echo -e "chr1\t100\t200\tgene1\nchr1\t1000\t1200\tgene2") -b <(echo -e "chr1\t50\t130\tgene3\nchr1\t150\t300\tgene4\nchr1\t500\t800\tgene5") -u
# Result: same as -wa, but duplicate rows are removed
chr1 100 200 gene18. Output only intervals in A that do not overlap B.
Parameter: -v (invert)
# Command
intersectBed -a <(echo -e "chr1\t100\t200\tgene1\nchr1\t1000\t1200\tgene2") -b <(echo -e "chr1\t50\t130\tgene3\nchr1\t150\t300\tgene4\nchr1\t500\t800\tgene5") -v
# Result
chr1 1000 1200 gene29. Filter by overlap fraction.
Parameters: -f -F (minimum overlap fraction)
# Command
intersectBed -a <(echo -e "chr1\t100\t200\tgene1\nchr1\t1000\t1200\tgene2") -b <(echo -e "chr1\t50\t130\tgene3\nchr1\t150\t300\tgene4\nchr1\t500\t800\tgene5") -f 0.5
# Result: only intervals where the overlap covers >=50% of A are output
chr1 150 200 gene1
# Command
intersectBed -a <(echo -e "chr1\t100\t200\tgene1\nchr1\t1000\t1200\tgene2") -b <(echo -e "chr1\t50\t130\tgene3\nchr1\t150\t300\tgene4\nchr1\t500\t800\tgene5") -F 0.35
# Result: only intervals where the overlap covers >=35% of B are output
chr1 100 130 gene110. Count how many times each interval in A overlaps B.
Parameter: -c (count overlaps)
# Command
intersectBed -a <(echo -e "chr1\t100\t200\tgene1\nchr1\t1000\t1200\tgene2") -b <(echo -e "chr1\t50\t130\tgene3\nchr1\t150\t300\tgene4\nchr1\t500\t800\tgene5") -c
# Result: each interval in A is output with the overlap count
chr1 100 200 gene1 2
chr1 1000 1200 gene2 0bedtools merge is used to merge overlapping or adjacent genomic intervals into continuous, non-overlapping intervals.
1. Merge overlapping and adjacent intervals.
# Command
bedtools merge -i <(echo -e "chr1\t50\t130\tgene3\nchr1\t100\t200\tgene1\nchr1\t150\t300\tgene4\nchr1\t500\t800\tgene5\nchr1\t1000\t1200\tgene2")
# Result
chr1 50 300
chr1 500 800
chr1 1000 12002. Set the maximum allowed gap length for merging.
Parameter: -d
# Command
bedtools merge -i <(echo -e "chr1\t50\t130\tgene3\nchr1\t100\t200\tgene1\nchr1\t150\t300\tgene4\nchr1\t500\t800\tgene5\nchr1\t1000\t1200\tgene2") -d 200
# Result: intervals with gaps <=200 bp are merged into one interval
chr1 50 12003. Preserve column information during merging.
Parameters: -c -o
# Command
bedtools merge -i <(echo -e "chr1\t50\t130\tgene3\nchr1\t100\t200\tgene1\nchr1\t150\t300\tgene4\nchr1\t500\t800\tgene5\nchr1\t1000\t1200\tgene2") -c 4 -o distinct
# The name column is preserved
chr1 50 300 gene1,gene3,gene4
chr1 500 800 gene5
chr1 1000 1200 gene2-c and -o must be used together. The common -o operations are listed below:
Usage:
4. Strand-specific merge.
Parameter: -s
# Command
bedtools merge -i <(echo -e "chr1\t50\t130\tgene3\t.\t+\nchr1\t100\t200\tgene1\t.\t+\nchr1\t150\t300\tgene4\t.\t-\nchr1\t500\t800\tgene5\t.\t+\nchr1\t1000\t1200\tgene2\t.\t+") -s -c 4,6 -o distinct
# Merge intervals by strand
chr1 50 200 gene1,gene3 +
chr1 150 300 gene4 -
chr1 500 800 gene5 +
chr1 1000 1200 gene2 +This tool can analyze overlaps across multiple BED files, identify genomic regions shared by different files, and report the coverage of each region across different files.
1. Basic usage.
# Command
bedtools multiinter -i <(echo -e "chr1\t100\t200\tgene1\nchr1\t1000\t1200\tgene2") <(echo -e "chr1\t50\t130\tgene3\nchr1\t150\t300\tgene4\nchr1\t500\t800\tgene5") -names A B
# Result
chr1 50 100 1 B 0 1
chr1 100 130 2 A,B 1 1
chr1 130 150 1 A 1 0
chr1 150 200 2 A,B 1 1
chr1 200 300 1 B 0 1
chr1 500 800 1 B 0 1
chr1 1000 1200 1 A 1 01. Default operation: remove regions from A that overlap B.
# Command
bedtools subtract -a <(echo -e "chr1\t100\t200\tgene1\nchr1\t1000\t1200\tgene2") -b <(echo -e "chr1\t50\t130\tgene3\nchr1\t150\t300\tgene4\nchr1\t500\t800\tgene5")
# Result
chr1 130 150 gene1
chr1 1000 1200 gene2